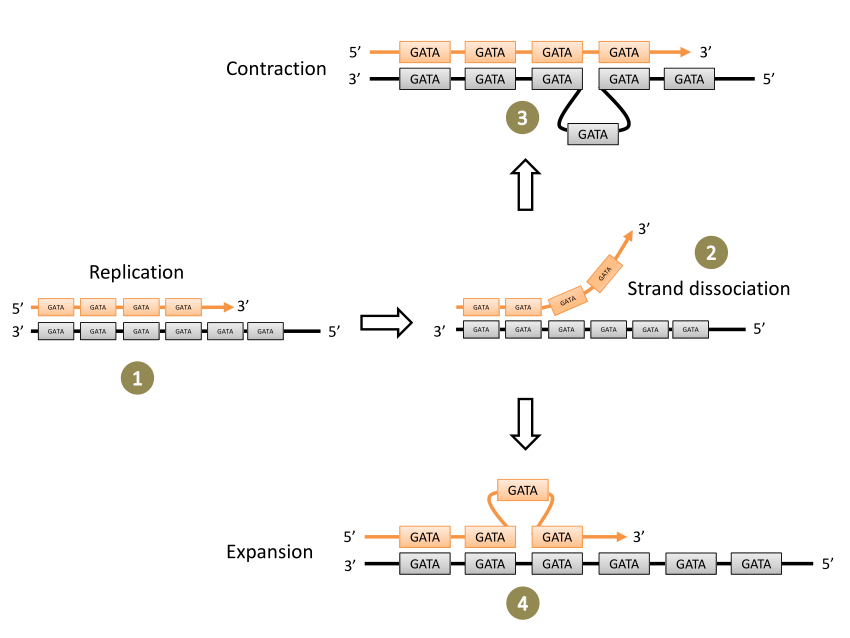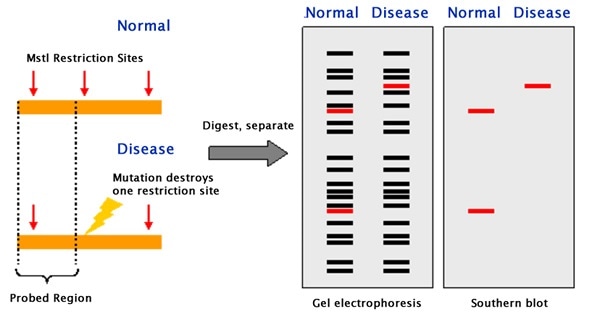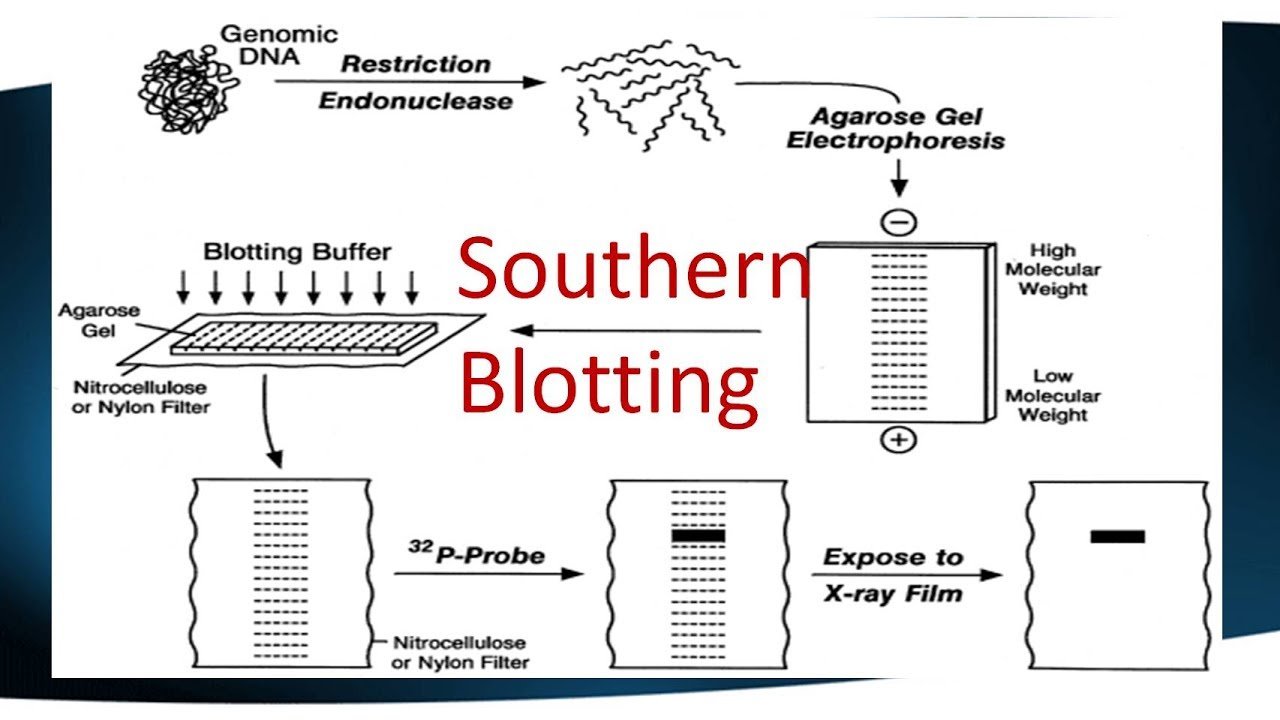
Introduction
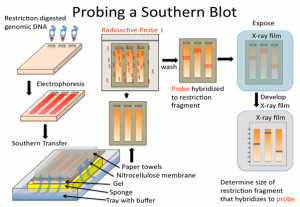
A Southern blot is a laboratory method used to detect specific DNA molecules among many other DNA molecules. The technique is named after its inventor, Edward Southern. As a laboratory procedure, Southern blots can be used to analyze an organism’s total DNA, also known as its genome, to identify a specific sequence of interest.
The first step in a Southern blot is to prepare the DNA mixture by breaking it into small fragments using a protein called a restriction enzyme. The mixture of DNA fragments is then separated by size using a technique called gel electrophoresis. After separation, the double-stranded DNA pieces denature or separate into single strands within the gel. The DNA is then transferred from the gel to a transfer membrane. Although this step is what gives the technique the name “southern transfer,” the term is generally used to describe the entire procedure.
Once the transfer is complete, the membrane carries all the bands originally on the gel. The membrane is then treated with a small piece of DNA or RNA called a probe, which has been designed to have a sequence that is complementary to a particular DNA sequence in the sample; this allows the probe to hybridize or bind to a specific DNA fragment on the membrane. Also, the probe has a label, which is usually a radioactive atom or a fluorescent dye. Thus, after hybridization, the probe allows the DNA fragment of interest to be detected among the many different DNA fragments on the membrane.
General procedure for drying
1. Sample homogenization involving DNA/RNA/protein purification that is performed after extraction from a variety of sources, such as cells or tissues.
2. Digest DNA with restriction enzymes into fragments, which is not necessary for RNA (northern blot).
3. Separation of the molecule of interest by membrane electrophoresis, generally on agarose gel for DNA fragments. In the case of RNA samples, they can be separated on an agarose gel in the presence of formaldehyde as a denaturing agent. This is necessary since formaldehyde limits the secondary structures of RNA molecules.
4. Transfer the molecules (DNA/RNA fragments) to a nitrocellulose membrane/nylon membrane from the gel.
5. Prehybridization (Blocking): Washing the nylon membrane with a prehybridization or blocking solution comprising salmon sperm DNA is required to block non-specific DNA interactions and also helps reduce background noise. Alternatively, there are some commercially available blocking buffers, such as PerfectHyb™ Plus buffer, in which salmon sperm DNA is not required for blocking purposes.
6. For probe preparation, a fresh 32P alpha-labelled dCTP-labeled DNA probe is prepared.
7. Hybridization or identification of the molecule that is achieved by incubating the blot with the specific labelled probe.
8. For detection of the probe and the DNA/RNA sequence of interest, the film is exposed to -80°C.
Beginning
Restriction endonucleases, which is enzymes, are used to break DNA into small fragments. These fragments are then separated by electrophoresis. The fragments obtained are then classified according to their size (kDa). Thus, the DNA fragments are transferred to blotting paper where they are incubated with probes. The probes used in Southern blotting can be very selective. They can selectively bind with a resolution of 1 in a million and the characteristics to bind to the intended target fragments.
Necessary materials reagents
- The buffer used for electrophoresis is TAE or TBE.
- The preferred agarose should be electrophoresis grade.
- For DNA staining, ethidium bromide (0.5 µg ml–1, dissolved in H2O) is used. But there are other DNA staining dyes, such as SYBR green, that can replace ethidium bromide for safe handling.
- 2X and 20X SSC (20X SSC composition includes 3.0 M NaCl and 0.3 M sodium citrate)
- 6X DNA Loading Buffer consisting of 0.25% Bromophenol Blue, 0.25% Xylene Cyanol FF, and 30% Glycerol in water.
- Suitable DNA markers of variable molecular weight, also called DNA ladders, are used as reference standards.
- The prehybridization and hybridization mixture consists of 0.5% SDS, 6X SSC, 5X Denhardt’s solution, and 100 mg ml–1 sheared denatured salmon sperm DNA of yeast tRNA.
- The widely used Denhardt solution for hybridization is composed of 0.02% Ficoll, 0.02% polyvinylpyrrolidone, and 0.02% bovine serum albumin (BSA)
- Paraffin oil
- Cellulose nitrate or nitrocellulose membrane filter with uniform porosity. (eg, Millipore 25 HAWP; nylon membranes used for blot protocols are available under various trade names from commercial vendors)
- RNase A (20 pg ml–1 in 2X SSC) is required for specific cleavage.
- Restriction enzyme and an appropriate buffer are used.
- Radioactively labelled RNA as a probe for specific detection where autoradiography is performed.
- For the detection of RNA labelled with labels such as 3H, 35S, 125I or 14C, 2,5-diphenyloxazole (PPO) in toluene at a concentration of 20% w/vol is required.
Team
1. Transfer of narrow strips of gel can be achieved using three pieces of glass or Plexiglas that are 5 cm × 20 cm in size with a thickness similar to the thickness of the gel.
2. There is a requirement for coarse, dry filter paper (four to five in number) or paper towels 10 cm × 18 cm in size.
3. The hybridization dish has a larger dimension (0.8 mm depth × 2 cm height × ~1 cm length) than the membrane used for hybridization, and the material used to develop the hybridization dish is Perspex (note: several alternative procedures for hybridization follow)
4. Four narrow pieces of Perspex that are similar in thickness to gel. Perspex length is sufficient to surround the gel at a distance of ~3 mm
5. A tray 20–50 mm (approx.) deep and 20 mm (approx.) in length and width larger than the gel
6. A sheet of glass long enough to fit in the pan and narrower to have a 10mm gap on each side
7. Several thick pieces of filter paper have a large area compared to the gel. The length of the filter paper is suitable to cover the glass sheet and can be immersed inside the tray.
8. A moistened piece of nitrocellulose membrane, which has a wider area that can cover the entire gel. The nitrocellulose membrane is placed on top of four strips of Perspex. Wetting of the nitrocellulose membrane is done using 2X SSC.
9. Paper towels that are stacked on top of each other.
10. Apparatus for straining gel.
11. A gel tank is necessary to perform electrophoresis.
12. Power supply is required for all device configurations.
Reagent Configuration
- DNA: The entire procedure begins using DNA digested with enzymes of varying concentrations that will quantify the optimal concentration of DNA and specify the restriction enzyme to be used. Generally, an amount of 1 µg of DNA derived from clones (eg, from plasmid or bacteriophage clones) is sufficient for plasmids having low copy numbers. We need larger amounts to carry out the separation of complex DNA (eg genomic DNA). The advisable range to consider would be 5 to 10 µg.
- The electrophoresis buffer used is TAE, which has a composition of 40 mM Tris, 20 mM acetic acid, 1 mM EDTA with a pH range of 7.4 to 8.2, which is normally prepared as a stock concentration of 20X or TBE composed of 89 mM Tris, 2.5 mM EDTA, 89 mM borate, normally made as a 10× stock)
- TAE is recommended to be better when running gels for a shorter time interval and when recovery of DNA fragments from the gel is to be carried out.
- TBE is considered to be a better buffer, especially when we have to run the gels for longer than 2 h.
Request
Southern blot is used in various applications. The main use of the Southern blot is to identify a specific DNA in a DNA sample. It is mainly used in the identification of viral infections and certain bacterial infections. In rDNA technology, the Southern blot technique is used to isolate a particular DNA. It is also useful in the study of genetic mutations and rearrangements, this property is used to diagnose neonatal diseases and genetic diseases. Due to the accuracy in DNA identification, this technique is used in phylogenetic studies, paternity and maternity analysis, forensic studies and personal identification.
Southern blotting can be applied to study the structure of a gene or to elucidate restriction enzyme maps. In particular, Southern blotting can be used to identify the methylated sites present for particular genes. This can be implemented by applying restriction nucleases such as MspI and HpaII, which can identify and cleave between identical sequences. The discovery of RFLPs by Southern blotting has aided in the mapping of several genomes that were crucial to map. In the field of immunology, clonal rearrangements of immunoglobulins, as well as T-cell receptor genes that play an important role in eliciting an immune response, can be analyzed by Southern blotting.